
Every empirical prescription makes two bets. The first is that the drug matches the organism and its resistance — that it will work on this patient, today. The second is quieter: that prescribing it will not push resistance a little further along. Culture settles the first two or three days later, by which point the prescription is written and the patient is either improving or not. The second is rarely settled at all — clinicians seldom learn what a course of antibiotics did to the resistance landscape it passed through.
What both bets share is that the information needed to inform them is not missing — it is in the sample, from the moment the swab or aspirate is taken. The problem has never been that the information does not exist, but that it cannot be read in time. The step that changes that is less visible than the sequencing: the ability to remove host DNA and recover the microbial signal carrying both pathogens and resistance genes.
Why antibiotic resistance is difficult to detect in clinical samples
The reason it cannot be read in time is sheer proportion. In most clinical samples the overwhelming majority of recoverable DNA is the patient's, not the microbe's — commonly 90 to 99 percent, and in some matrices well beyond — and the resistance genes sit in the small remainder. Sequence the sample as it arrives and the instrument spends almost all its capacity reading the human genome. Resistance is not absent from the sample; it is hidden in it.
The hidden problem: host DNA and lost signal in metagenomic sequencing
Removing host DNA before sequencing is the critical step that enables clinical metagenomics to detect pathogens and antibiotic resistance genes in real time. Salt-active nucleases — the enzymes HL-SAN and M-SAN HQ — digest host DNA under the high-salt conditions selective-lysis protocols use, where conventional nucleases lose activity; the methods built around them were developed by the academic and clinical groups whose work follows.
The effect is direct: Yang et al. (2019) reported that depletion raised the microbial share of reads in lower respiratory samples from roughly one percent to approximately 48 percent — and whatever you are looking for in that fraction, from a pathogen's resistance genes to the resistome of a whole community, becomes nearly fifty times easier to see.
The first bet: does the drug fit
The first bet is about the patient in front of you, and here depletion buys speed: with most of the host DNA gone, far fewer reads are needed to reach the pathogen and the resistance genes it carries, and the answer arrives while it can still change the prescription.
Bellankimath et al. (2025), in Nature Communications, showed this on complicated urinary tract infections: complete pathogen identification and resistance profiling in approximately four hours, using an optimised protocol built on M-SAN HQ — helped by the high bacterial load of complicated UTI, enough to sequence directly without pre-amplification.
Alcolea-Medina, Snell et al. (2025), in The Lancet Microbe, ran a unified protocol — detecting bacteria, viruses and fungi together, using HL-SAN to deplete host DNA — on respiratory samples from critically ill patients at Guy's and St Thomas' NHS hospitals. It detected pathogens routine testing had missed and, the part that matters, its results informed antibiotic adjustments in patients already in intensive care.
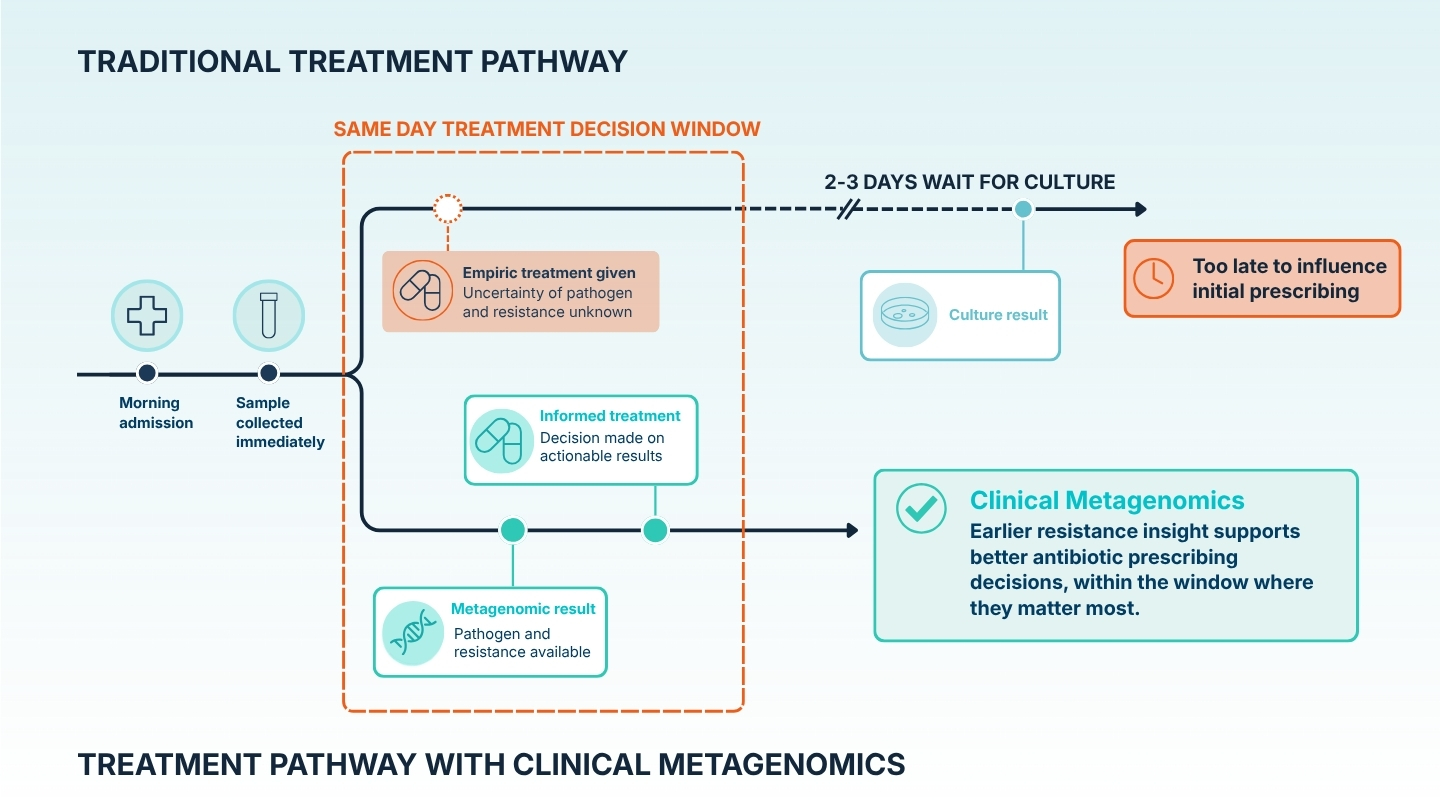
The matrices differ and so does the tempo, but the point holds across both: resistance information actionable for the prescribing decision arrived before culture could have produced it — not replacing culture, but knowing something culture cannot tell you yet, while the decision is still open.
The second bet: what does antibiotic use leave behind in the resistome?
The second bet is harder, because it cannot be settled one prescription at a time. No single test tells a clinician whether today's course nudged the resistance landscape. What can be studied instead is how different prescribing strategies, applied over time, leave different marks on the resistome.
Rofael et al. (2023) illustrate the distinction, and the shared matrix makes it easy to miss. Their samples are also respiratory — but the resemblance to the ICU work ends there. Where the ICU study diagnoses acute infection in critically ill patients, Rofael followed stable patients with bronchiectasis, a chronic condition involving repeated antibiotic courses, watching how the resistance genes in their airways behaved over time under three regimens: continuous prophylaxis, antibiotics only during flare-ups, and neither. Same site, same chemistry, an entirely different clinical question.
Depletion matters here for a different reason than in the acute case. There, the resistance you care about belongs to a dominant pathogen; here it is spread across the whole community, much of it in low-abundance organisms, so a measure of how the resistome shifts is only as trustworthy as the depletion that lifted those rare genes into view.
Using HL-SAN-based metagenomics, Rofael found a trend: patients who used antibiotics during flare-ups carried the widest variety of resistance genes; those on continuous prophylaxis carried modestly more than those on no antibiotics at all. These are trends from a pilot of 17 patients, not statistically significant findings, and the authors are explicit that they need testing in larger cohorts before clinical conclusions follow.
The result is not actionable for any single prescription. Its value is slower: the start of an evidence base that could, in time, tell stewardship which prescribing strategies leave resistance better or worse off — the second bet informed at the level of policy, not the individual decision.
One step, two bets: how host DNA depletion changes both decisions
The two bets are different in kind — one about this patient now, the other about the resistance landscape over time — and usually addressed by different means. What connects them is the same buried resistome and the same step that exposes it: remove the host DNA, and resistance becomes visible — quickly enough to inform the prescription in hand, completely enough to study what prescribing does over months.
One chemistry, both timescales. The same depletion step has been validated across blood, wound, stool, and more than twenty sample types in total, so the stewardship argument made here for UTI and lung is not confined to those matrices.
A comprehensive review of what this chemistry delivers across clinical settings and more than twenty sample types — its limits weighed as carefully as its strengths — is coming later this year.
For the clinical detail behind the ICU evidence above, the webinar with Adela Alcolea-Medina and Luke Snell sets out how the unified protocol is run in practice.
And for how this chemistry came to exist, Justin O'Grady tells the origin story in his interview How an accidental error helped unlock clinical metagenomics on the ArcticZymes website.
References
- Alcolea-Medina A, Snell L, Humayun G et al. Rapid pan-microbial metagenomics for pathogen detection and personalised therapy in the intensive care unit: a single-centre prospective observational study. The Lancet Microbe, 2025; 6. https://doi.org/10.1016/j.lanmic.2025.101174
- Bellankimath, A.B., Branders, S., Kegel, I. et al. Metagenomic sequencing enables accurate pathogen and antimicrobial susceptibility profiling in complicated UTIs in approximately four hours. Nat Commun 17, 187 (2026). https://doi.org/10.1038/s41467-025-66865-8
- Rofael SAD, Brown J, Lipman MCI, Lowe DM, Spratt D, Quaderi S, et al. Impact of prophylactic and ‘rescue pack’ antibiotics on the airway microbiome in chronic lung disease. BMJ Open Respiratory Research. 2023;10:e001335. https://doi.org/10.1136/bmjresp-2022-001335
- Yang, L., Haidar, G., Zia, H. et al. Metagenomic identification of severe pneumonia pathogens in mechanically-ventilated patients: a feasibility and clinical validity study. Respir Res 20, 265 (2019). https://doi.org/10.1186/s12931-019-1218-4



